Promising tool to analyze sequences
Sequence Analysis
To extract knowledge
-
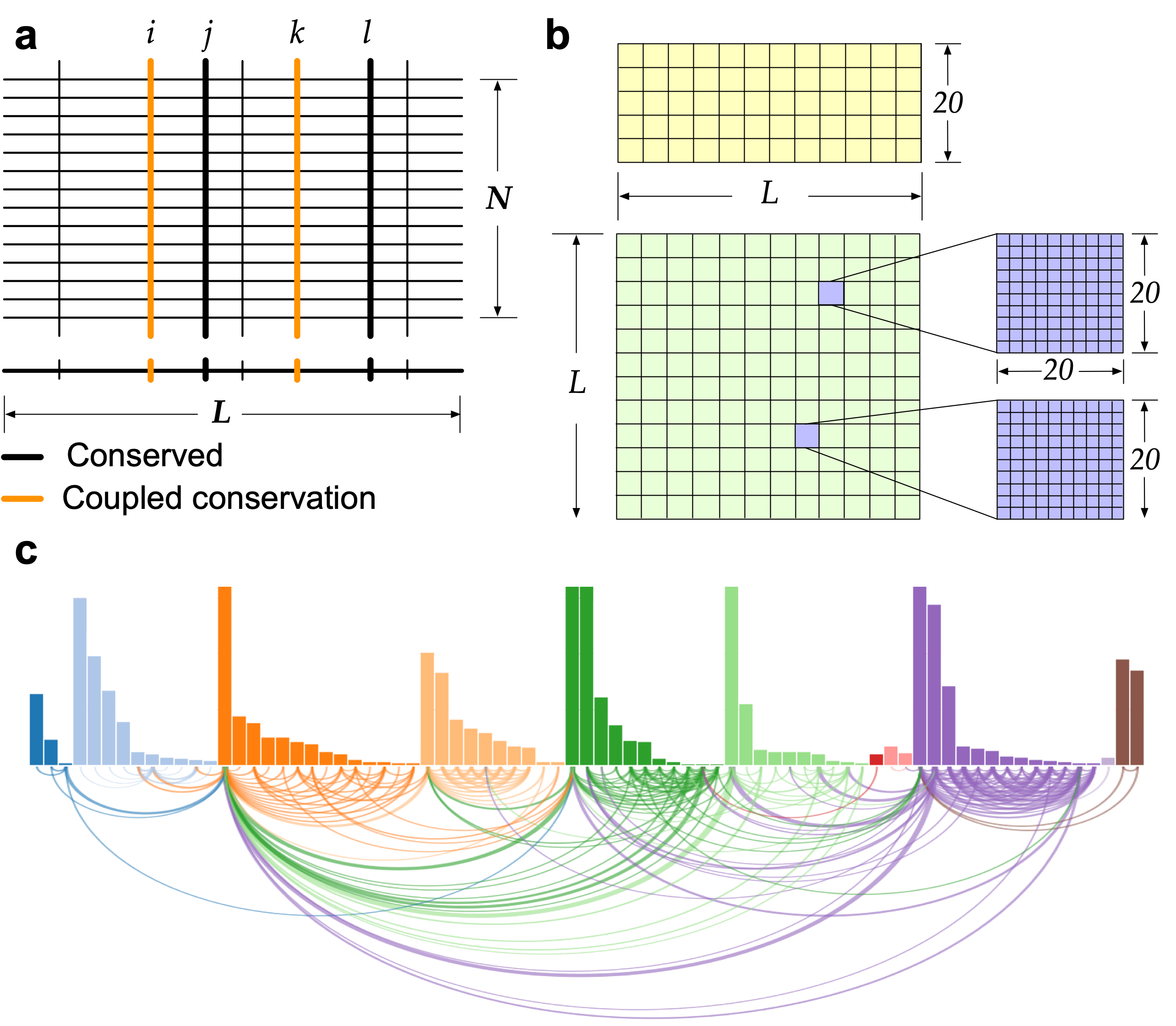
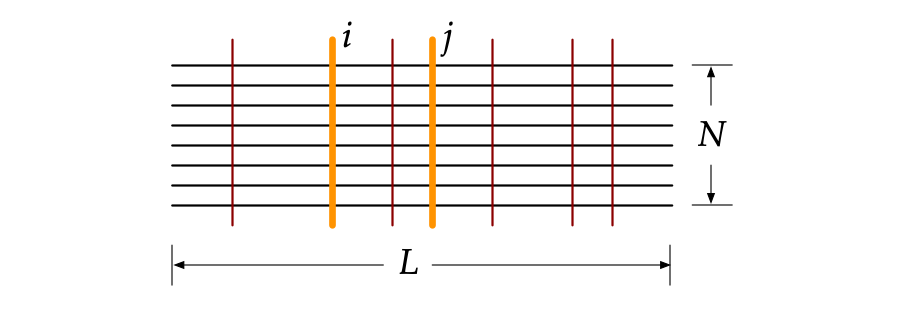
Naturally co-occurring amino acids, term coevolution, in a protein family play a significant role in both protein engineering and folding, and it is expanding in recent years from the studies of the effects of single-site mutations to the complete re-design of a protein and its folding, especially in structure prediction. An approach (SAEC) is developed to identify the evolutionary signatures (highly ordered networks of coupled amino acids, termed "residue communities", RCs) from protein homologous sequences.