Welcome!
We research and build powerful general-purpose algorithms to accelerate biology, advance therapeutics, and benefit humanity. Leveraging physics-informed algorithms, we seek to understand the fundamental principles evolved for protein structure and function, to encode these principles into computer programs, and to use them to address some of the most complex and interesting challenges in biology.
Recent posts
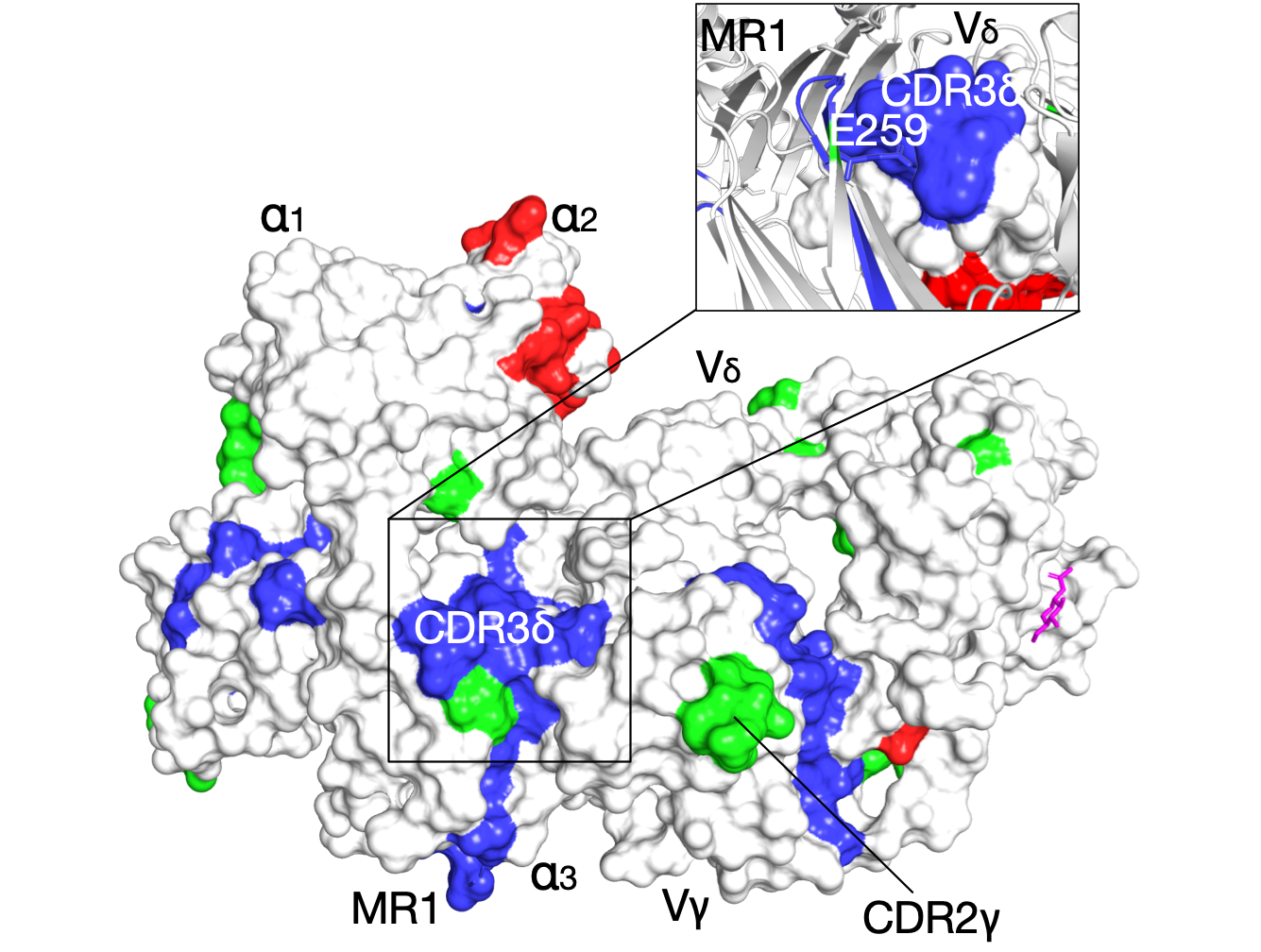
The evolutionary signatures of γδ T-Cell receptor
Naturally co-occurring amino acids (coevolution), are essential in protein engineering and folding, spanning research from single-site mutations to complete protein redesign, particularly in predicting 3D structures. We computationally analyze evolutionary couplings from extensive sequences, uncovering critical signatures for specific molecular interactions. Applied to the G7 γδ T-cell receptor, this identifies key residues for its unique binding mode. The analysis aligns with known functional sites, facilitating receptor docking under the MR1 antigen presenting groove. Moreover, we observe that inter-residue contacts closely correlate with receptor activation states, paving the way to connect protein sequence with function, without requiring structural data or precise biological activity measurements.

Residue communities identify the tunnel in the Lipophilic Envelope-spanning Tunnel B (LetB)
LetB forms a periplasm-spanning tunnel consisting of seven stacked rings and provides a hydrophobic pathway for the translocation of lipids across the periplasm. Rings 1, 5, 6, and 7 are dynamic and adopt open and closed states. Our method has successfully identified functional communities in the LetB (PDB: 6V0C) as shown in the above figure. The pore-facing residues in red are inferred using Amoai and contribute to the formation of the tunnel that mediates lipid transport in different states.