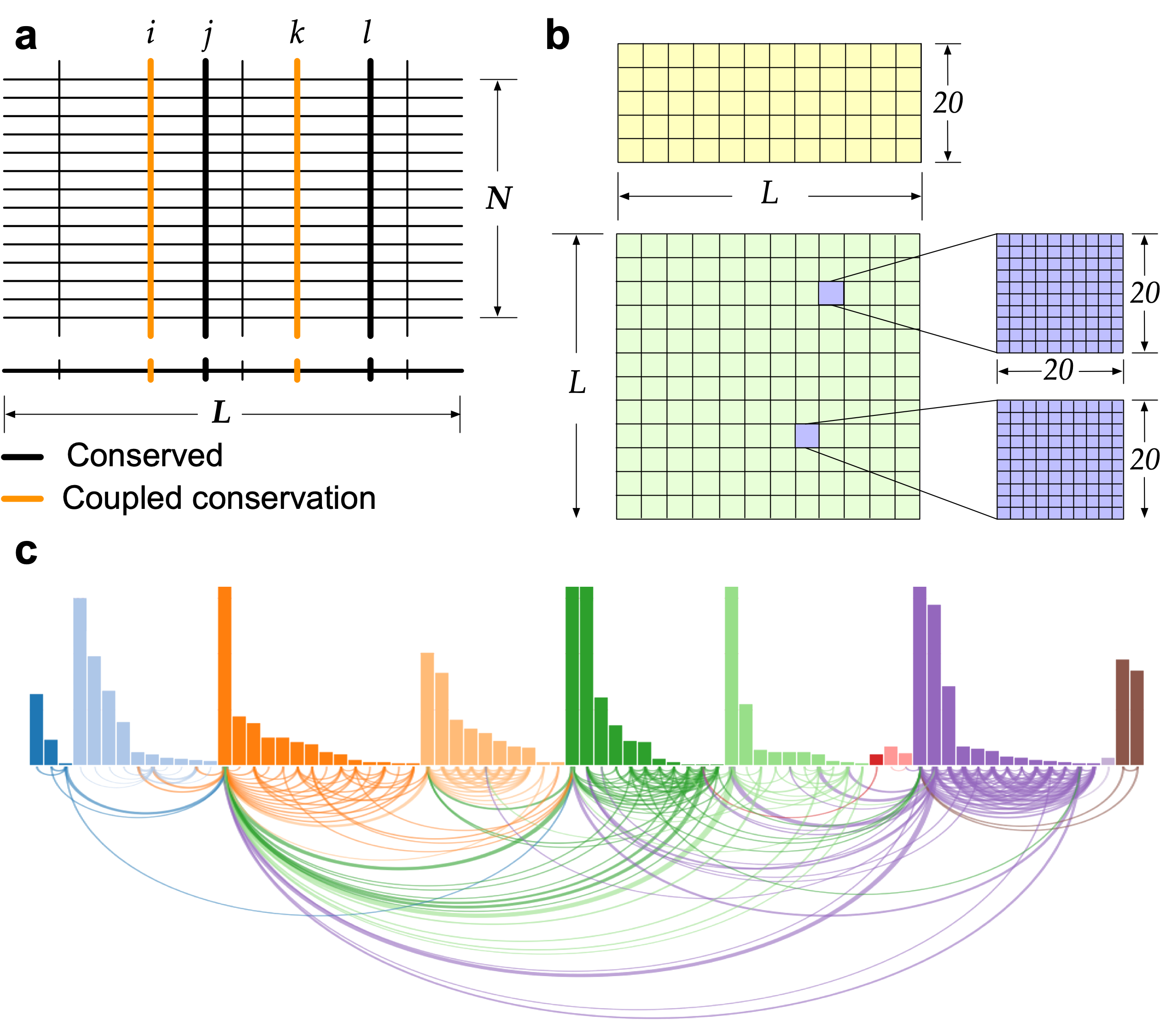
Evolutionary Signatures
Naturally co-occurring amino acids, term coevolution, in a protein family play a significant role in both protein engineering and folding, and it is expanding in recent years from the studies of the effects of single-site mutations to the complete re-design of a protein and its folding, especially in structure prediction. An approach (SAEC) is developed to identify the evolutionary signatures (highly ordered networks of coupled amino acids, termed "residue communities", RCs) from protein homologous sequences.
Identification of evolutionary signatures
How to identify evolutionary signatures?
Evolutionary information is conserved in naturally occurring proteins, and it plays a significant role in specifying their three-dimensional structure and function. An approach (SAEC) is developed to identify the evolutionary signatures (highly ordered networks of coupled amino acids, termed "residue communities", RCs) from protein sequence profiles.
Missense effects: quantifying the pathogenicity of genetic variants in human disease-related genes has the potential to advance evidence-based clinical decision making and transform healthcare. We are developing physics-/evolution-informed machine learning approaches to address the knowledge gap.
Multiple sequence alignment
How to generate an MSA?
Given a protein amino acid sequence, its multiple sequence alignment (MSA) is obtained from two databases by the profiles HMM1 and HHblits2 homology search tools. Firstly, an alignment of a target protein is obtained by searching its sequence against the Uniclust30 database using HHblits with the default parameters of an E-value threshold 0.001. The other alignment of the same protein is generated by the default five search iterations of jackhmmer in the HMM suite, searching the query sequence against the UniRef90 database. Finally, the two alignments are combined and aligned according to the target sequence. The final alignment of each protein is combined from the two obtained alignments according to the query sequence. Thereafter, it is trimmed based on minimum coverage, which satisfies two basic rules3,4: (1) a single site with more than 90% gaps across the MSA will be removed; and (2) a sequence with the percentage of gaps less than a given threshold (80%) will be deleted from the MSA. The weight, ω, of each sequence is computed using Leri 4.
References:
- S. R. Eddy, PLoS computational biology 7(10), e1002195 (2011)
- M. Mirdita et al., Nucleic acids research 45, D170–D176 (2017)
- N. J. Cheung, W. Yu. BMC bioinformatics 20, 1–11 (2019)
- N. J. Cheung et al., Comput. Struct. Biotechnol. J. (2021)
- S. Kullback, R. A. Leibler, The annals mathematical statistics 22,79–86 (1951).
Degree of conservation
How nature conserves info.?
The Kullback-Leibler relative entropy5 is used to measure how different the observed amino acid A at the ith position would be if A randomly occurred with an expected probability distribution.
For discrete probability distributions \(P\) and \(Q\) defined on the same sample space, \( \mathcal{X} \), the relative entropy from \(Q\) to \(P\) is defined6 to be $$D_{KL}(P||Q)=\sum_{x\in\mathcal{X}}P(x)\log\left(\frac{P(x)}{Q(x)}\right),$$ where is equivalent to $$D_{KL}(P||Q)=-\sum_{x\in\mathcal{X}}P(x)\log\left(\frac{Q(x)}{P(x)}\right).$$ In other words, it is the expectation of the logarithmic difference between the probabilities \(P\) and \(Q\), where the expectation is taken using the probabilities \(P\). Relative entropy is only defined in this way if, for all \(x\), \( Q(x)=0 \) implies \(P(x)=0\) (absolute continuity). Otherwise, it is often defined as \(+\infty\), but the value \(+\infty\) is possible even if \(Q(x)\neq 0\) everywhere5, provided \(\mathcal {X}\) is infinite.
References:
- S. Kullback, R. A. Leibler, The annals mathematical statistics 22,79–86 (1951).
- David J.C., MacKay, Information theory, inference, and learning algorithms (First ed.). Cambridge University Press. p. 34. 22,79–86 (1951).
Evolutionary couplings
What are evolutionary couplings?
A pairwise coupling is a pattern between two covariant amino acids, and those couplings are conserved across evolution due to the functional and structural constraints. They can be used to predict protein structures, binding interfaces and protein-protein interactions, as well as protein design.
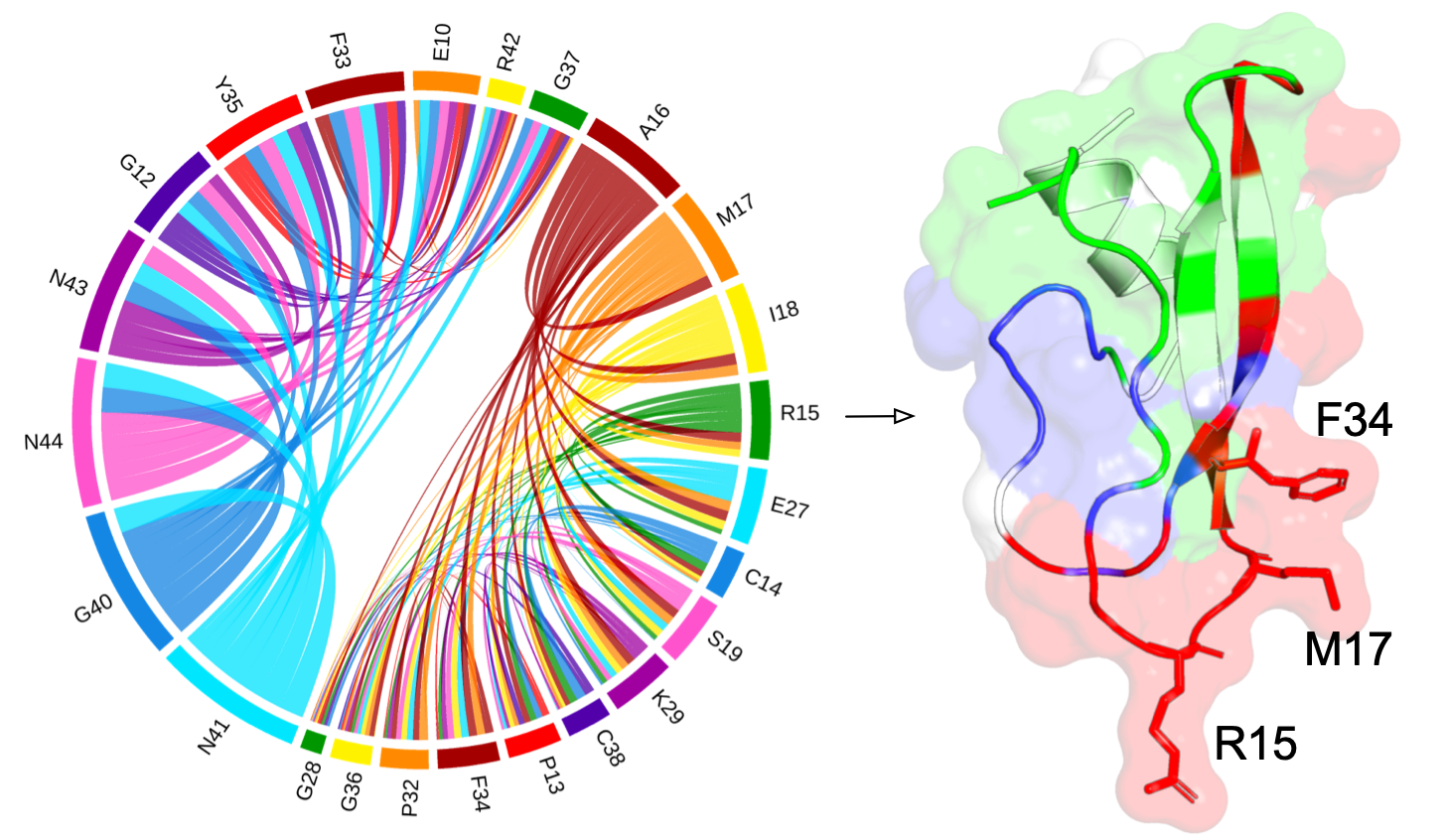
Residue communities
What are residue communities?
The couplings between pairwise amino acids are analyzed using the principles in the SAEC method1 to make the residue communities as much statistically independent as possible. The principles are as follows:
- community I consists of residues at the \(i\)th position of \(\mathbf{v}_{k=2}^i>\text{max}(\mathbf{v}_{k=4}^i, \epsilon)\),
- community II includes residues at the \(i\)th position of \(\mathbf{v}_{k=2}^i<-\text{max}(\mathbf{v}_{k=4}^i, \epsilon)\),
- community III includes residues at the \(i\)th position of \(\mathbf{v}_{k=4}^i>\text{max}(\mathbf{v}_{k=2}^i, \epsilon)\).
We use an \(\epsilon=0.05\) as a threshold to project the amino acids reduced from the coupling matrix and extract meaningful residue communities.
Mapping to structure
Where are coupled residues located?
A pairwise coupling is a pattern between two covariant amino acids, and those couplings are conserved across evolution due to the functional and structural constraints. They can be used to predict protein structures, binding interfaces and protein-protein interactions, as well as protein design.
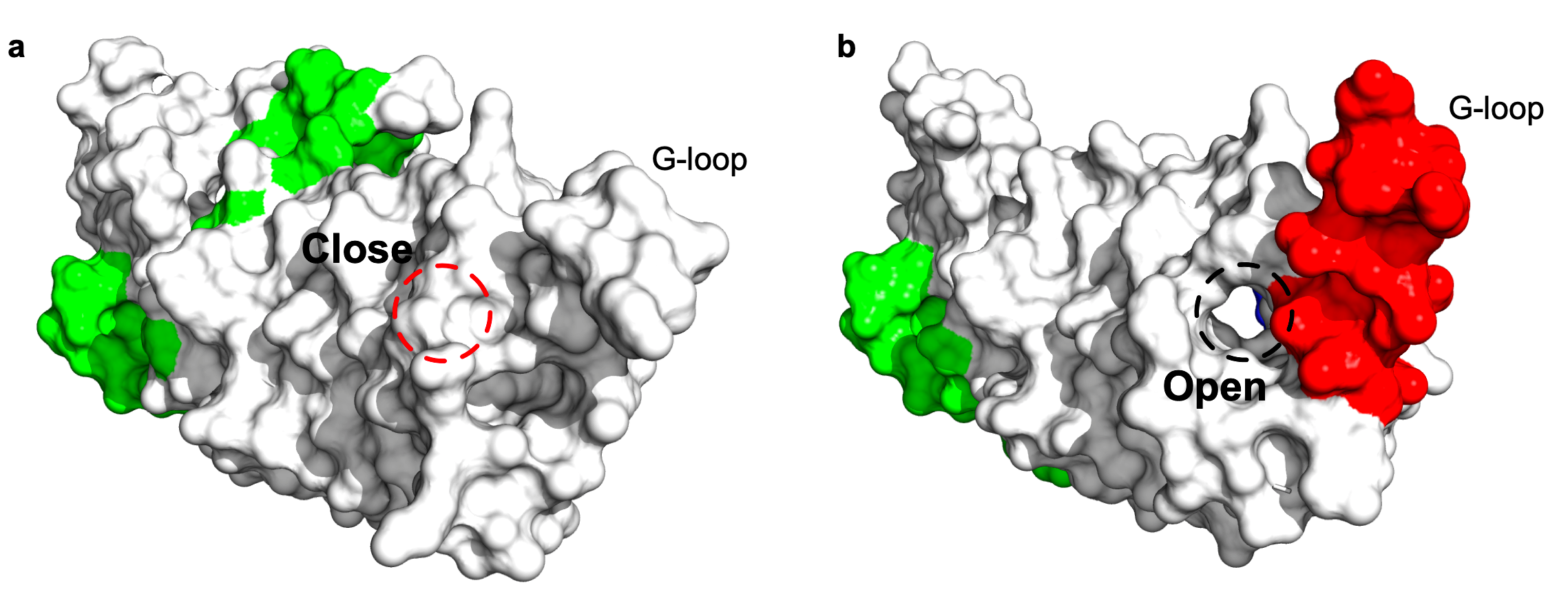
Function-relevant residues
How residue communities are relevant to protein function?
A conformational change of the G-loop make an extended structure to plug into the head region of the scMdm12Δ and cover the solvent-exposed concave surface of scMdm12Δ2. The SAEC method reflects that residue community (in red) is relevant to the conformational change, the two structures of Mmm1 as single domain and in the complex are aligned to each other for comparison. The statistical qualities of the residue communities (in green) do not capture the important information from the G-loop when Mmm1 adopts a shape as a monomer, while the Mmm1 changes its shape via the G-loop (residue community in red) in response to the formation of a complex between the Mmm1 and Mdm12 proteins.